The following publications have used the ABACUS software. Publications that only mentioned the ABACUS will not be included below.
We encourage explicitly mentioning ABACUS with proper citations in your publications, so we can more easily find and list these publications.
Last update date: November 28, 2024
2024
Efficient Homojunction Tin Perovskite Solar Cells Enabled by Gradient Germanium Doping
Zhenzhu Zhao, Mulin Sun, Yuyang Ji, Kaitian Mao, Zongming Huang, Chengjian Yuan, Yuqian Yang, Honghe Ding, Yingguo Yang, Yu Li, Wenjing Chen, Junfa Zhu, Jing Wei, Jixian Xu, Watcharaphol Paritmongkol, Antonio Abate, Zhengguo Xiao, Lixin He, Qin Hu
Nano Lett., 2024, 24 (18), 5513–5520.
DOI: 10.1021/acs.nanolett.4c00646
Tuning of Berry-curvature dipole in TaAs slabs: An effective route to enhance the nonlinear Hall response
Hongsheng Pang, Gan Jin, Lixin He
Phys, Rev, Mater., 2024, 8 (4), 43403.
DOI: 10.1103/PhysRevMaterials.8.043403
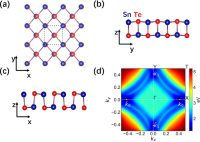
Peculiar band geometry induced giant shift current in ferroelectric SnTe monolayer
Gan Jin, Lixin He
npj Comput. Mater, 2024, 10 (1), 23.
DOI: 10.1038/s41524-024-01213-w
Simultaneous Catalytic Acceleration of White Phosphorus Polymerization and Red Phosphorus Potassiation for High-Performance Potassium-Ion Batteries
Hai Yang, Fuxiang He, Fanfan Liu, Zhefei Sun, Yu Shao, Lixin He, Qiaobao Zhang, Yan Yu
Adv. Mater. (Deerfield Beach Fla,), 2024, 36 (3), e2306512.
DOI: 10.1002/adma.202306512
Ab initio electronic structure calculations based on numerical atomic orbitals: Basic fomalisms and recent progresses
Peize Lin, Xinguo Ren, Xiaohui Liu, Lixin He
Wires Comput. Mol Sci, 2024, 14 (1).
DOI: 10.1002/wcms.1687
Ultrafast shift current dynamics in WS2 monolayer
Fuxiang He, Daqiang Chen, Xinguo Ren, Sheng Meng, Lixin He
Phys, Rev, Res., 2024, 6 (1), 13123.
DOI: 10.1103/PhysRevResearch.6.013123
2023
PYATB: An efficient Python package for electronic structure calculations using ab initio tight-binding model
Gan Jin, Hongsheng Pang, Yuyang Ji, Zujian Dai, Lixin He
Comput. Phys. Commun., 2023, 291, 108844.
DOI: 10.1016/j.cpc.2023.108844
Interplay between magnetic structures and surface states in MnBi2Te4 from first-principles studies
Zujian Dai, Gan Jin, Lixin He
Phys. Rev. B, 2023, 108 (8), 85112.
DOI: 10.1103/PhysRevB.108.085112
Implementation of the meta-GGA exchange-correlation functional in numerical atomic orbital basis: With systematic testing on SCAN, rSCAN, and r2SCAN functionals
Renxi Liu, Daye Zheng, Xinyuan Liang, Xinguo Ren, Mohan Chen, Wenfei Li
J. Chem. Phys., 2023, 159 (7), 74109.
DOI: 10.1063/5.0160726
2022
Reproducibility of Hybrid Density Functional Calculations for Equation-of-State Properties and Band Gaps
Yuyang Ji, Peize Lin, Xinguo Ren, Lixin He
J. Phys. Chem., A, 2022, 126 (35), 5924–5931.
DOI: 10.1021/acs.jpca.2c05170
Real-Time, Time-Dependent Density Functional Theory Study on Photoinduced Isomerizations of Azobenzene Under a Light Field
Fuxiang He, Xinguo Ren, Jun Jiang, Guozhen Zhang, Lixin He
J. Phys. Chem. Lett., 2022, 13 (2), 427–432.
DOI: 10.1021/acs.jpclett.1c03442
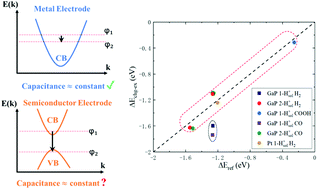
A caveat of the charge-extrapolation scheme for modeling electrochemical reactions on semiconductor surfaces: an issue induced by a discontinuous Fermi level change
Yu Liu, Xinlong Ding, Mohan Chen, Shenzhen Xu
Phys. Chem. Chem. Phys., 2022, 24 (25), 15511–15521.
DOI: 10.1039/D2CP00642A
Disordered hyperuniform quasi-one-dimensional materials
Duyu Chen, Yu Liu, Yu Zheng, Houlong Zhuang, Mohan Chen, Yang Jiao
Phys. Rev. B, 2022, 106 (23), 235427.
DOI: 10.1103/PhysRevB.106.235427
First-principles calculations of the surface states of doped and alloyed topological materials via band unfolding method
Zujian Dai, Gan Jin, Lixin He
Comput. Mater. Sci., 2022, 213, 111656.
DOI: 10.1016/j.commatsci.2022.111656
Plane-wave-based stochastic-deterministic density functional theory for extended systems
Qianrui Liu, Mohan Chen
Phys. Rev. B, 2022, 106 (12), 125132.
DOI: 10.1103/PhysRevB.106.125132
Density Functional Theory Plus Dynamical Mean Field Theory within the Framework of Linear Combination of Numerical Atomic Orbitals: Formulation and Benchmarks
Xin Qu, Peng Xu, Rusong Li, Gang Li, Lixin He, Xinguo Ren
J. Chem. Theory Comput., 2022, 18 (9), 5589–5606.
DOI: 10.1021/acs.jctc.2c00472
DFT+U within the framework of linear combination of numerical atomic orbitals
Xin Qu, Peng Xu, Hong Jiang, Lixin He, Xinguo Ren
J. Chem. Phys., 2022, 156 (23), 234104.
DOI: 10.1063/5.0090122
2021
Design Principles of Sodium/Potassium Protection Layer for High-Power High-Energy Sodium/Potassium-Metal Batteries in Carbonate Electrolytes: a Case Study of Na2 Te/K2 Te
Hai Yang, Fuxiang He, Menghao Li, Fanyang Huang, Zhihao Chen, Pengcheng Shi, Fanfan Liu, Yu Jiang, Lixin He, Meng Gu, Yan Yu
Adv. Mater. (Deerfield Beach Fla,), 2021, 33 (48), e2106353.
DOI: 10.1002/adma.202106353
Strategy for constructing compact numerical atomic orbital basis sets by incorporating the gradients of reference wavefunctions
Peize Lin, Xinguo Ren, Lixin He
Phys. Rev. B, 2021, 103 (23), 235131.
DOI: 10.1103/PhysRevB.103.235131
Efficient Hybrid Density Functional Calculations for Large Periodic Systems Using Numerical Atomic Orbitals
Peize Lin, Xinguo Ren, Lixin He
J. Chem. Theory Comput., 2021, 17 (1), 222–239.
DOI: 10.1021/acs.jctc.0c00960
Accurate stress calculations based on numerical atomic orbital bases: Implementation and benchmarks
Daye Zheng, Xinguo Ren, Lixin He
Comput. Phys. Commun., 2021, 267, 108043.
DOI: 10.1016/j.cpc.2021.108043
Peculiar diffusion behavior of AlCl4 intercalated in graphite from nanosecond-long molecular dynamics simulations*
Qianpeng Wang, Daye Zheng, Lixin He, Xinguo Ren
Chin. Phys, B, 2021, 30 (10), 107102.
DOI: 10.1088/1674-1056/ac0692
Beryllium and Magnesium Metal Clusters: New Globally Stable Structures and G0W0 Calculations
Sunila Bakhsh, Xiaohui Liu, Yanyong Wang, Lixin He, Xinguo Ren
J. Phys. Chem., A, 2021, 125 (7), 1424–1435.
DOI: 10.1021/acs.jpca.0c08960
Copper-doped beryllium and beryllium oxide interface: A first- principles study
Yu Liu, Xiaohui Liu, Mohan Chen
J. Nucl. Mater., 2021, 545, 152733.
DOI: 10.1016/j.jnucmat.2020.152733
Retention and recycling of deuterium in liquid lithium-tin slab studied by first-principles molecular dynamics
Daye Zheng, Zhen-Xiong Shen, Mohan Chen, Xinguo Ren, Lixin He
J. Nucl. Mater., 2021, 543, 152542.
DOI: 10.1016/j.jnucmat.2020.152542
2020
Accuracy of Localized Resolution of the Identity in Periodic Hybrid Functional Calculations with Numerical Atomic Orbitals
Peize Lin, Xinguo Ren, Lixin He
J. Phys. Chem. Lett., 2020, 11 (8), 3082–3088.
DOI: 10.1021/acs.jpclett.0c00481
First-principles study of magnon-phonon interactions in gadolinium iron garnet
Lian-Wei Wang, Li-Shan Xie, Peng-Xiang Xu, Ke Xia
Phys. Rev. B, 2020, 101 (16), 165137.
DOI: 10.1103/PhysRevB.101.165137
Optimizing the Void Size of Yolk-Shell Bi@Void@C Nanospheres for High- Power-Density Sodium-Ion Batteries
Hai Yang, Lin-Wei Chen, Fuxiang He, Jiaqing Zhang, Yuezhan Feng, Lukang Zhao, Bin Wang, Lixin He, Qiaobao Zhang, Yan Yu
Nano Lett., 2020, 20 (1), 758–767.
DOI: 10.1021/acs.nanolett.9b04829
Disordered hyperuniformity in two-dimensional amorphous silica
Yu Zheng, Lei Liu, Hanqing Nan, Zhen-Xiong Shen, Ge Zhang, Duyu Chen, Lixin He, Wenxiang Xu, Mohan Chen, Yang Jiao, Houlong Zhuang
Sci. Adv., 2020, 6 (16), eaba0826.
DOI: 10.1126/sciadv.aba0826
2019
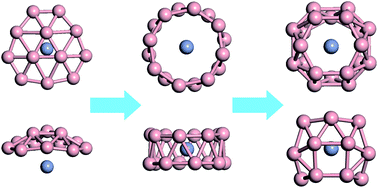
Structure evolution of chromium-doped boron clusters: toward the formation of endohedral boron cages
Xuecheng Shao, Xin Qu, Siyu Liu, Lihua Yang, Jinghai Yang, Xiaohui Liu, Xin Zhong, Shuai Sun, G. Vaitheeswaran, Jian Lv
Rsc Adv., 2019, 9 (5), 2870–2876.
DOI: 10.1039/C8RA09143A
A DFT study of energetic and structural properties of a full turn of A-form DNA under relaxed and stretching conditions
Yue Liu, Xinguo Ren, Lixin He
J. Chem. Phys., 2019, 151 (21), 215102.
DOI: 10.1063/1.5129716
Cooperative Effect in a Graphite Intercalation Compound: Enhanced Mobility of AlCl4 in the Graphite Cathode of Aluminum-Ion Batteries
Qianpeng Wang, Daye Zheng, Lixin He, Xinguo Ren
Phys, Rev, Appl., 2019, 12 (4), 44060.
DOI: 10.1103/PhysRevApplied.12.044060
2018
Diffusion coefficients of Mg isotopes in MgSiO3 and Mg2SiO4 melts calculated by first-principles molecular dynamics simulations
Xiaohui Liu, Yuhan Qi, Daye Zheng, Chen Zhou, Lixin He, Fang Huang
Geochim. Cosmochim. Acta, 2018, 223, 364–376.
DOI: 10.1016/j.gca.2017.12.007
2017
First-principles calculations and model analysis of plasmon excitations in graphene and graphene/hBN heterostructure
Pengfei Li, Xinguo Ren, Lixin He
Phys. Rev. B, 2017, 96 (16), 165417.
DOI: 10.1103/PhysRevB.96.165417
First-principles molecular dynamics study of deuterium diffusion in liquid tin
Xiaohui Liu, Daye Zheng, Xinguo Ren, Lixin He, Mohan Chen
J. Chem. Phys., 2017, 147 (6), 64505.
DOI: 10.1063/1.4997635
2015
Large-scale ab initio simulations based on systematically improvable atomic basis
Pengfei Li, Xiaohui Liu, Mohan Chen, Peize Lin, Xinguo Ren, Lin Lin, Chao Yang, Lixin He
arXiv, 2015, 1503.00097.
DOI: 10.48550/arXiv.1503.00097
2013
Accelerating atomic orbital-based electronic structure calculation via pole expansion and selected inversion
Lin Lin, Mohan Chen, Chao Yang, Lixin He
J. Phys., Condens. Matter: Inst. Phys. J., 2013, 25 (29), 295501.
DOI: 10.1088/0953-8984/25/29/295501
2011
Electronic structure interpolation via atomic orbitals
Mohan Chen, G-C Guo, Lixin He
J. Phys., Condens. Matter: Inst. Phys. J., 2011, 23 (32), 325501.
DOI: 10.1088/0953-8984/23/32/325501
2010
Systematically improvable optimized atomic basis sets for ab initio calculations
Mohan Chen, G-C Guo, Lixin He
J. Phys., Condens. Matter: Inst. Phys. J., 2010, 22 (44), 445501.
DOI: 10.1088/0953-8984/22/44/445501